Практикум 4. Расчет атомных орбиталей водорода¶
import numpy as np
import scipy.special
import scipy.misc
import numpy2cube # Автор: Андрей Демкив
Зададим волновую фунцию
def w(n,l,m,d):
x,y,z = np.mgrid[-d:d:30j,-d:d:30j,-d:d:30j] # Зададим сетку из 30 точек от -d до d
# Переход от декартовых к сферическим координатам
r = lambda x,y,z: np.sqrt(x**2+y**2+z**2) # расстояние от начала координат
theta = lambda x,y,z: np.arccos(z/r(x,y,z)) # зенитный угол
phi = lambda x,y,z: np.arctan(y/x) # азимутальный угол
a0 = 1
R = lambda r,n,l: (2*r/n/a0)**l * np.exp(-r/n/a0) * \
scipy.special.genlaguerre(n-l-1,2*l+1)(2*r/n/a0) # Радиальная компонента волновой функции
WF = lambda r,theta,phi,n,l,m: R(r,n,l) * \
scipy.special.sph_harm(m,l,phi,theta) # Волновая функция (радиальная часть * угловую)
absWF = lambda r,theta,phi,n,l,m: np.absolute(WF(r,theta,phi,n,l,m))**2 # Квадрат модуля волновой функции
return WF(r(x,y,z),theta(x,y,z),phi(x,y,z),n,l,m)
Посчитаем орбитали для первых трех главных квантовых чисел
# Зададим цикл по перебору квантовых чисел
for n in range(1,4):
for l in range(0,n):
for m in range(-l,l+1,1):
d = 10 * n
step=2*d/(30-1)
grid=w(n, l, m, d)
name='%s-%s-%s' % (n,l,m)
# для сохранения нужно задать координаты старта grid и шаг по каждому направлению
numpy2cube.npy2cube(grid, (-d,)*3, (step,)*3, name+'.cube')
Нарисуем их в паймоле
import __main__
__main__.pymol_argv = [ 'pymol', '-x' ]
import pymol
pymol.finish_launching()
from pymol import cmd,stored
cmd.reinitialize()
#ramp is defined as: [x1, rgb1, alpha1, ...]
cmd.volume_ramp_new('my_ramp', [\
-0.8, 1.00, 0.00, 0.00, 0.12,
-0.01, 1.00, 0.75, 0.00, 0.88,
-0.005, 1.00, 0.00, 0.00, 0.00,
0, 1,1,1,0.0,
0.005, 0.00, 0.00, 1.00, 0.00,
0.01, 0.00, 0.75, 1.00, 0.88,
0.8, 0.00, 0.00, 1.00, 0.12])
import time
clip_vals = [0.001,
-25, 0, -25, 0,
-37, 0, -35, 0, 0, 0, 0.001, 0, 0]
clip_i = 0
for n in range(1,4):
for l in range(0,n):
for m in range(-l,l+1,1):
name='%s-%s-%s' % (n,l,m)
cmd.hide("volume", "all")
cmd.load(f"{name}.cube")
cmd.volume(f"{name}_volume", name)
cmd.volume_color(f"{name}_volume", "my_ramp")
cmd.clip("near", clip_vals[clip_i])
if clip_vals[clip_i] == 0:
cmd.set_view ([\
0.999755025, 0.011685935, 0.018848220,\
-0.011722559, 0.999928892, 0.001830711,\
-0.018824987, -0.002051400, 0.999820948,\
0.000000000, 0.000000000, -242.648696899,\
0.000000000, 0.000000000, 0.000000000,\
-48.560935974, 533.858337402, -20.000000000])
else:
# Чтобы красиво видеть pz и dz^2 орбитали
cmd.set_view([\
-0.018926797, -0.164738640, -0.986155510,\
-0.008280928, 0.986323476, -0.164607957,\
0.999786615, 0.005050731, -0.020031987,\
0.000000000, 0.000000000, -181.888442993,\
0.000000000, 0.000000000, 0.000000000,\
181.388442993, 182.388442993, -20.000000000])
cmd.draw()
time.sleep(2)
cmd.png(f"{name}.png")
cmd.clip("near", -clip_vals[clip_i])
clip_i += 1
Посмотрим на них. Из-за разных set_view масштаб у нас разный. Он один и тот же между группой s, pz и dz^2 орбиталей, а также между всеми остальными орбиталями. В ряде случаев орбитали "срезаны", чтобы увидеть плотность в плоскости экрана, близкой к ядру.
from IPython.display import display,Image
for n in range(1,4):
for l in range(0,n):
for m in range(-l,l+1,1):
display(Image(f"{n}-{l}-{m}.png"))













Сравним с тем, что рисует нам ORCA (2s-2p орбитали)
file = open("h.inp", "w")
orca_inp = \
"""! UHF SVP XYZFile
%plots
Format CUBE
MO("H1.cube",1,0);
MO("H2.cube",2,0);
MO("H3.cube",3,0);
MO("H4.cube",4,0);
end
*xyz 0 2
H 0 0 0
*"""
file.write(orca_inp)
file.close()
cmd.reinitialize()
cmd.volume_ramp_new('my_ramp', [\
-0.8, 1.00, 0.00, 0.00, 0.12,
-0.01, 1.00, 0.75, 0.00, 0.88,
-0.005, 1.00, 0.00, 0.00, 0.00,
0, 1,1,1,0.0,
0.005, 0.00, 0.00, 1.00, 0.00,
0.01, 0.00, 0.75, 1.00, 0.88,
0.8, 0.00, 0.00, 1.00, 0.12])
for i in range(1, 5):
name=f"H-{i}"
cmd.hide("volume", "all")
cmd.load(f"{name}.cube")
cmd.volume(f"{name}_volume", name)
cmd.volume_color(f"{name}_volume", "my_ramp")
time.sleep(2)
cmd.png(f"{name}.png")
display(Image(f"{name}.png"))




Видим отличие в 2p орбиталях. Отличие, вероятно, объясняется тем, что наш способ расчета является "честным", а ORCA по умолчанию немного хитрит и считает не 2px, 2py, а (2px + 2py)/2, (2px - 2py)/2j вместо них, так называемые "real orbitals". Почитать про них можно, например, в параграфе 13.7.2 книжки Molecular Modelling for Beginners 2nd edition.
Несмотря на то, что мы рисуем вроде как "честные" орбитали, при визуализации в cube файл попадает только вещественная часть волновой функции. Этим объясняется "странный" вид орбиталей с l>0 и |m|>0, ведь они имеют ненулевую комлексную компоненту. Правильная визуализация с помощью программы hydrogen приводится ниже в конце практикума, но ее в виде cube-файла не сохранить, так как для каждого узла, в котором мы сохраняем значение комплексной функции, надо сохранить не одно число, а 2: модуль (или квадрат модуля) и фазу волновой функции.
Примечательно, что ORCA посчитала нам только орбитали 0-4 (мы при этом нарисовали только 2s и 2p, всего 4 штуки, а не 5). Думаю, это объясняется тем, что в базисе SVP для атома водорода всего 5 базисных функций: большая s, малая s и 3 поляризационных p. Соответственно, из них можно родить 2 орбитали s-типа и 3 орбитали p-типа. Вот и получили наши 5 функций.
Возьмем базис намного побольше (aug-cc-pV5Z), чтобы получить орбитали энергией повыше, и посмотрим, что ORCA нам нарисует. Нарисовали орбитали 2s-4s (14 штук).
cmd.reinitialize()
cmd.volume_ramp_new('my_ramp', [\
-0.8, 1.00, 0.00, 0.00, 0.12,
-0.01, 1.00, 0.75, 0.00, 0.88,
-0.005, 1.00, 0.00, 0.00, 0.00,
0, 1,1,1,0.0,
0.005, 0.00, 0.00, 1.00, 0.00,
0.01, 0.00, 0.75, 1.00, 0.88,
0.8, 0.00, 0.00, 1.00, 0.12])
for i in range(1, 15):
name=f"H{i}"
cmd.hide("volume", "all")
cmd.load(f"aug-cc-pV5Z/{name}.cube")
cmd.volume(f"{name}_volume", name)
cmd.volume_color(f"{name}_volume", "my_ramp")
time.sleep(2)
cmd.png(f"aug-cc-pV5Z/{name}.png")
display(Image(f"{name}.png"))














Здесь снова видим отличие в p-орбиталях. С увеличением базиса, появились и орбитали с энергией повыше (d). d-орбитали тоже отличаются от тех, что нарисовали мы. Все они похожи на dz^2. Думаю, объясняется это из тех же соображений, что и в случае p-орбиталей: имеем дело с real orbitals.
Помимо книжки выше, можно на википедии почитать про real orbitals. То же самое. Еще там гифка красивая есть.
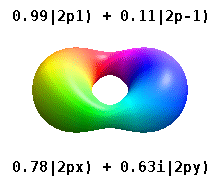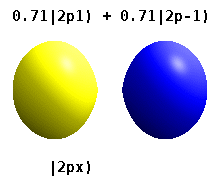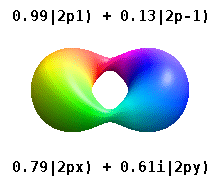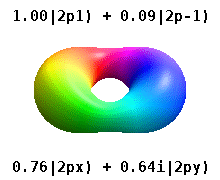
Еще одна картинка с википедии. https://upload.wikimedia.org/wikipedia/commons/2/2a/Atomic_orbitals_spdf_m-eigenstates_and_superpositions.png
В левой части рисунка вещественные орбитали, в правой - честные решения уравнения Шредингера, цветом обозначена комплексная фаза волновой функции
